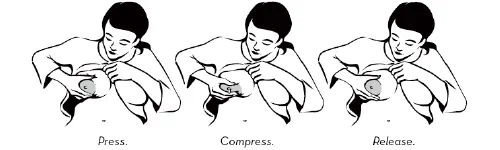
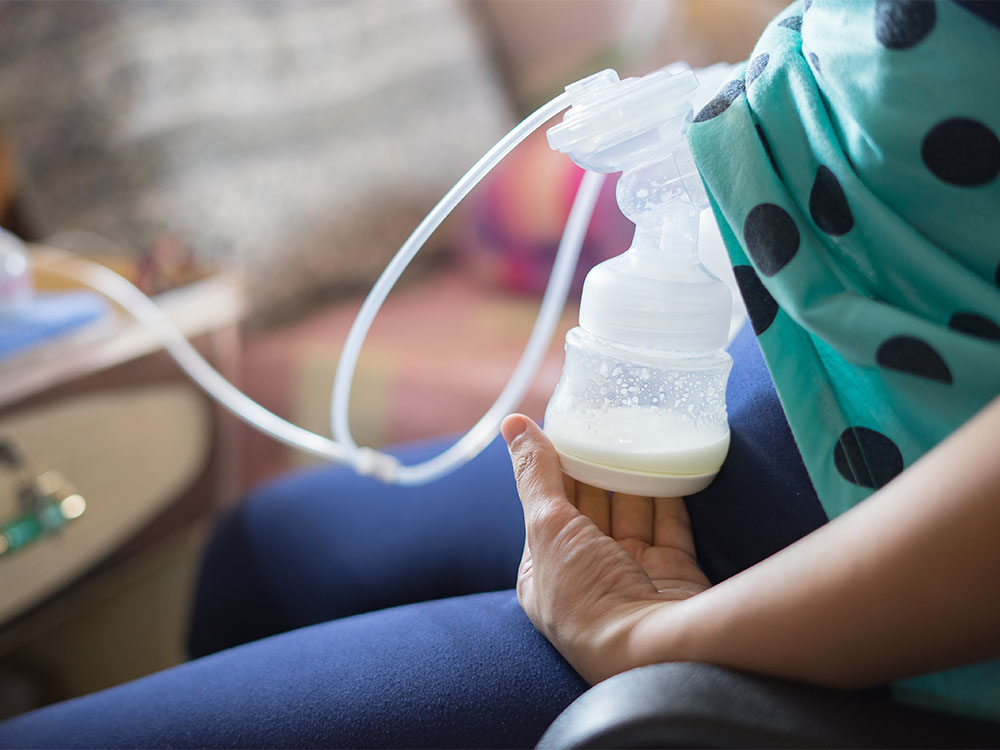
Proper storage of expressed breast milk (EBM) helps preserve its nutritional and immunological properties for later feeding when the mother is away.
2. Containers for Storage
Use clean, sterilized glass or BPA-free plastic containers with tight-fitting lids.
Breast milk storage bags (food-grade, pre-sterilized) are convenient for freezing.
Avoid ordinary plastic bottles or disposable liners.
3. Labeling
Each container should be labeled with:
Date and time of expression
Baby’s name (if used in hospital or daycare)
4. Storage Guidelines
Location
Temperature
Duration
Remarks
Room temperature
Up to 25°C (77°F)
4–6 hours
Keep in a cool, clean area away from direct sunlight
Refrigerator (back portion)
2–4°C (35–40°F)
Up to 72 hours (3 days)
Do not store in refrigerator door due to temperature fluctuation
Freezer (separate door)
–18°C or lower
Up to 3–6 months
Keep in small portions; leave space for expansion
Deep freezer
–20°C
Up to 6–12 months
Best for long-term storage
Insulated cooler box with ice packs
~15°C
Up to 24 hours
For temporary transport or travel
5. Thawing and Warming
Thaw in refrigerator overnight or by placing the container in warm water (<37°C).
Do not microwave or boil — destroys antibodies and nutrients.
Swirl gently (do not shake) to mix separated fat layers.
Once thawed, use within 24 hours and do not refreeze.
6. Hygiene
Wash hands before expressing or handling milk.
Use clean pump parts and containers for each session.
Avoid touching the inside of lids or bottles.
7. Key Points
Always use oldest milk first (“first in, first out”).
Discard leftover milk from feeding bottle after use.
Observe for odor or curdling — discard if spoiled.
Summary:
Safe breast milk storage requires hygienic handling, correct temperature, and appropriate duration. Properly stored milk retains most of its nutritional, immunological, and protective properties, ensuring safe feeding for infants when direct breastfeeding isn’t possible.
An enlarged placenta isn’t usually a reason to panic. The word is big (placentomegaly), but most of the time it doesn’t cause problems.
Some conditions play a role. A larger placenta may be linked to hypertension, anemia, or diabetes — but your doctor will monitor these and the health of your baby.
Your baby’s growth matters most. Even if your placenta measures big, steady fetal development is the important goal, so keep up your regular appointments to be sure everything’s right on track.
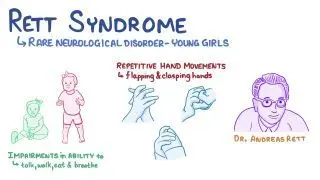
Rett syndrome is a neurodevelopmental disorder that primarily affects girls, characterized by normal early development followed by regression of acquired skills, especially speech and purposeful hand movements, with onset typically between 6–18 months of age.
Etiology
Genetic cause: Mutation in MECP2 gene (methyl-CpG-binding protein 2) on the X chromosome (Xq28)
Inheritance: Usually sporadic (de novo); rarely familial
Pathophysiology: Dysfunction of MECP2 protein → abnormal brain maturation and synaptic development
Epidemiology
Affects 1 in 10,000–15,000 female births
Lethal in males (most do not survive infancy unless mosaic or XXY)
Clinical Features
Phases of Disease
Early Onset (6–18 months)
Normal development initially
Gradual loss of interest in surroundings
Loss of purposeful hand skills
Deceleration of head growth (acquired microcephaly)
Rapid Destructive Phase (1–4 years)
Loss of speech and purposeful hand use
Stereotyped hand movements: hand-wringing, washing, clapping, or mouthing
Gait ataxia, truncal apraxia
Autistic-like behavior
Plateau Phase (2–10 years)
Some improvement in social interaction and eye contact
Hypothyroidism is a clinical state resulting from deficiency of thyroid hormone production or action, leading to a generalized slowing of metabolic processes.
It may be:
Congenital (Neonatal) – present at birth.
Acquired (Childhood) – develops later due to autoimmune, iatrogenic, or other causes.
2. Classification
A. Based on Level of Defect
Type
Site of Defect
TSH
T4/T3
Primary
Thyroid gland
↑
↓
Secondary
Pituitary
↓/N
↓
Tertiary
Hypothalamus
↓/N
↓
Peripheral (Resistance)
Target tissue
N/↑
N/↑
B. Based on Onset
Congenital hypothyroidism (CH)
Acquired hypothyroidism
3. Epidemiology
CH: ~1 in 2,000–4,000 live births.
More common in females.
Acquired form common in older children/adolescents, often autoimmune (Hashimoto’s).
thyroid gland
4. Etiology
A. Congenital Hypothyroidism
Thyroid dysgenesis (80–85%)
Agenesis, ectopy, or hypoplasia.
Usually sporadic.
Dyshormonogenesis (10–15%)
Inborn errors of thyroid hormone synthesis (autosomal recessive).
Leishmaniasis — MD Pediatrics Note (Based on Nelson Textbook of Pediatrics)
Table of Contents(toc)
Introduction
Leishmaniasis is a spectrum of protozoal diseases caused by Leishmania species, transmitted by the bite of infected female phlebotomine sandflies.
cutaneous leishmaniasis
Disease manifestations depend on the species involved and the host immune response.
Major clinical forms:
Visceral leishmaniasis (VL / kala-azar)
Cutaneous leishmaniasis (CL)
Mucocutaneous leishmaniasis (MCL)
Etiology and Classification
Form
Causative Species
Geographic Distribution
Visceral
L. donovani, L. infantum (chagasi)
South Asia, East Africa, Latin America
Cutaneous
L. tropica, L. major, L. mexicana, L. braziliensis
Middle East, Africa, Americas
Mucocutaneous
L. braziliensis complex
Central & South America
Epidemiology
Endemic in >80 countries; affects poor, rural populations.
Vectors:Phlebotomus (Old World), Lutzomyia (New World).
Reservoirs: Humans (L. donovani), dogs, rodents.
Transmission: Sandfly bite, rarely congenital or via transfusion.
Phlebotomus
Pathogenesis
Inoculation of promastigotes → engulfed by macrophages → transform into amastigotes → intracellular multiplication → spread to RES (liver, spleen, bone marrow).
Disease severity depends on cell-mediated immunity (CMI).
Category: Inherited bone marrow failure syndrome (IBMFS) Inheritance: Autosomal recessive (rarely X-linked) Gene defects: >22 genes identified (FANCA, FANCC, FANCG most common) → defective DNA interstrand crosslink repair.
fanconi anemia notes
1. Pathophysiology
Defect in DNA repair (Fanconi/BRCA pathway) → chromosomal breakage and hypersensitivity to DNA cross-linking agents (e.g., mitomycin C, diepoxybutane).
Progressive bone marrow failure (due to stem cell depletion) and genomic instability → predisposition to malignancies.
Multisystem developmental abnormalities due to impaired cell proliferation during embryogenesis.
2. Epidemiology
Incidence: ~1 in 100,000–250,000 live births.
Carrier frequency: ~1 in 200.
Median age of diagnosis: 7–9 years.
~90% develop marrow failure by age 40.
3. Clinical Features
A. Hematologic
Pancytopenia (usually first manifests with thrombocytopenia or macrocytic anemia).
हामी प्रायः रगत परीक्षणका लागि हातको नसाबाट (vein) रगत निकालिन्छ भन्ने कुरा जान्दछौं। तर कहिलेकाहीँ स्वास्थ्यकर्मीले नाडीबाट (artery) पनि रगत निकाल्छन्। यो सामान्य रगत परीक्षणभन्दा फरक र विशिष्ट उद्देश्यका लागि गरिन्छ।
ABG sampling technique why and when
नाडीबाट रगत निकाल्नुको मुख्य कारण — “Arterial Blood Gas (ABG)” परीक्षण
नाडीबाट रगत निकाल्ने मुख्य उद्देश्य Arterial Blood Gas (ABG) test हो।
यो परीक्षणले शरीरमा रहेका अक्सिजन (O₂), कार्बन डाइअक्साइड (CO₂) र रगतको अम्ल–क्षार (pH) सन्तुलन कस्तो छ भन्ने देखाउँछ।
यो जानकारी फोक्सो र मुटुको कार्य कस्तो छ भन्ने बुझ्न अत्यन्त जरुरी हुन्छ।
यो परीक्षण कहिले गरिन्छ ?
जब बिरामीलाई अक्सिजन कमी (hypoxia) को शंका हुन्छ।
सास फेर्न गाह्रो भएको अवस्थामा (जस्तै– दमा, COPD, pneumonia, ARDS)।
भेन्टिलेटरमा राखिएका बिरामीहरूमा, अक्सिजनको मात्रा ठिक छ कि छैन भनेर हेर्न।
ABG गर्नुअघि प्रायः Allen’s test गरिन्छ, जसले हातको रक्तप्रवाह सुरक्षित छ कि छैन भन्ने पक्का गर्छ।
कसरी निकालिन्छ ?
बिरामीलाई आराम दिन्छ।
छालालाई सफा गरिन्छ (antiseptic)।
नाडीको धड्कन भेटाएर सुई प्रयोग गरी सिधै नाडीभित्र सुई प्रवेश गरिन्छ।
रगत सिधै syringe मा स्वचालित रूपमा भरिन्छ, किनकि नाडीको दबाब (pressure) बढी हुन्छ।
त्यसपछि तुरुन्तै syringe लाई बर्फमा राखी ल्याबमा पठाइन्छ ताकि ग्यासहरू नबदलिऊन्।
नसाबाट होइन, नाडीबाट किन ?
नसाको रगतले शरीरको अक्सिजन र कार्बन डाइअक्साइडको सन्तुलन सही रूपमा देखाउँदैन, किनभने त्यो पहिले नै ऊतकहरूबाट फर्किएको हुन्छ।
तर नाडीको रगत भने फोक्सोबाट निस्किएको ताजा अक्सिजनयुक्त रगत हो, जसले शरीरको साँच्चिकै ग्यास स्थिति जनाउँछ।
त्यसैले फोक्सो, सासफेर्ने प्रणाली वा अक्सिजन थेरापी मूल्याङ्कन गर्न नाडीबाट रगत आवश्यक पर्छ।
के जोखिम हुन्छ ?
सामान्यतया सुरक्षित भए पनि केही साइड इफेक्ट हुन सक्छन् —
नाडीमा दबाबको कारण दुखाइ वा निलो दाग (bruise)
कहिलेकाहीँ रगत बग्ने वा clot बन्ने समस्या
धेरै पटक सुई लगाउँदा नाडीको क्षति वा हात सुन्निनु
त्यसैले यो परीक्षण प्रशिक्षित स्वास्थ्यकर्मी (जस्तै चिकित्सक वा नर्स) ले मात्र गर्नुपर्छ।
सारांशमा
नाडीबाट रगत निकाल्नु साधारण परीक्षण होइन, तर अत्यन्त महत्त्वपूर्ण चिकित्सकीय प्रक्रिया हो जसले शरीरको अक्सिजन, कार्बन डाइअक्साइड र अम्ल–क्षार सन्तुलनबारे सटीक जानकारी दिन्छ।
यसले चिकित्सकलाई बिरामीको सासफेर्ने स्थिति बुझ्न, भेन्टिलेटर मिलाउन, र उपचारको प्रभाव मूल्याङ्कन गर्न मद्दत गर्छ।
Stay Connected with Dr. Chaitanya Joshi, MD
YouTube Channel
Watch health videos, tips, and updates from Dr. Chaitanya MD.

























")





