Croup is an acute viral inflammatory disease of the upper airway involving the larynx, trachea, and bronchi, leading to subglottic edema and airway obstruction.
Epidemiology
Age: 6 months – 3 years (can occur up to 6 years)
Male > Female
Peak: Autumn & early winter
Usually preceded by URTI
Etiology
Viral (most common)
Parainfluenza virus type 1 (most common)
Parainfluenza 2 & 3
RSV
Influenza A & B
Adenovirus
Human metapneumovirus
Rare bacterial causes
Mycoplasma
Secondary bacterial infection (uncommon)
Pathophysiology
Viral infection → inflammation & edema of subglottic region
Subglottis is the narrowest part of pediatric airway
Small edema → marked increase in airway resistance
Leads to inspiratory stridor & respiratory distress
What are the causes, symptoms, complications and treatment of vitamin D deficiency?
Contents
Table of Contents(toc)
vitamin d capsules
What is vitamin D?….
What are sources of vitamin D?…………
What is the function of vitamin D in our body? Why do we need vitamin D?…………
What is the daily requirement of vitamin D?………….
What causes vitamin D deficiency?……………
What are symptoms of vitamin D deficiency?…………
What are the complications of vitamin D deficiency?………..
How to prevent vitamin D deficiency?……..
How to treat vitamin D deficiency?….
How to test vitamin D in our body?…..
What is the dose of vitamin D supplementation?…..
Do we overdose on vitamin D?………..
What should I do if I think I lack vitamin D?……
What is vitamin D?
Vitamin D is an essential micronutrient which can be found in various food sources. Vitamin D is fat-soluble, and few foods naturally have vitamin D. Except fatty fish liver, other foods are poor sources of vitamin D. Vitamin D is needed for bone metabolism and calcium balance in the body.
Synthesis of vitamin D in skin is the main source of vitamin D for humans. The vitamin D in the skin is formed by exposure of Ultraviolet light into the skin, converting 7-dehydrocholesterol to provitamin D3. This is then converted to cholecalciferol by temperature dependent rearrangement. The sun exposure to face and arms only produces up to 200 International units per day of vitamin D.
Fatty fish liver is another major source. Minor sources include milk, meat and animal liver, eggs, some vegetables and mushrooms.
vitamin d and bone
What is the function of vitamin D in our body? Why do we need vitamin D?
After production in skin, or after taking vitamin D (D2 or D3 from food), our blood is converted to 25-hydroxyvitamin D and then to 1,25-hydroxyvitamin D in the kidney. This is the active form of vitamin D. The functions of vitamin D are:
Prevention form heart disease like hypertension and heart attack
Prevention from other endocrine diseases and diabetes
Boosting immune system
Helping brain development and prevention of cognitive function decline
Prevention from mental illnesses
What is the daily requirement of vitamin D?
Recommended dietary allowance RDA of vitamin D is as follows:
Up to 12 months of age: 400 IU per day (=10 mcg)
Children 1-18 years, people up to 70 years: 600 IU (=15mcg) per day
People above 70 years: 800 IU (=20mcg) per day
People often have low Vitamin D intake, and most people are deficient in vitamin D. Many people are at considerable risk for deficiency. Thus, it’s recommended for regular supplement of vitamin D to all the high-risk populations. Now a days milk fortification with vitamin D has also started to meet this requirement.
People with malabsorption disorders require high dose supplementation of vitamin D as high as 40000 IU per day.
What causes vitamin D deficiency?
Worldwide, billions of people lack vitamin D. Some factors mentioned below may cause vitamin D deficiency or resistance in our body:
Low exposure to sunlight
Low dietary intake
Low fat intake
Malabsorption disorders or syndromes
Residence in regions where there is low sun exposure
Impaired ability of body to use inactive vitamin D (liver or renal dysfunction)
Resistance of body to act to the vitamin D present in our body
Older age
High dose of steroids drug intake
vitamin d deficiency depiction
What are symptoms of vitamin D deficiency?
Most people are asymptomatic initially
Bone pain and tenderness
Muscle weakness
Fractures
Difficulty walking
What are the complications of vitamin D deficiency?
Increases bone loss
Osteopenia and osteoporosis
Hypocalcemia
Hypophosphatemia
Secondary hyperparathyroidism
Phosphaturia
Osteomalacia
Muscle weakness, cancers, decreased immunity or increased autoimmune diseases, asthma, hypertension, MI, diabetes, bad pregnancy outcomes
How to prevent vitamin D deficiency?
Get adequate exposure to direct sunlight, especially in the morning (10 am to 2 am) time when there is adequate concentration and band of UV light in sunlight
Eat fish and fish liver that have vitamin D (cod, salmon, swordfish, tuna)
Serum vitamin D (25-hydroxyvitamin D) level can be measured by blood test to confirm or screen for vitamin D deficiency. The common consensus is that 30 ng/mL (nanogram per milliliter) or 75 nmol/L is sufficient for most individuals. However, the reference range may vary depending on the population and consensus.
Serum PTH (parathyroid hormone) level is inversely related to serum vitamin D level so it can also be measured to check for vitamin D deficiency.
What is the dose of vitamin D supplementation?
Despite adequate dietary and behavioral measures to prevent vitamin D deficiency, people may have deficiency and may even have clinical manifestations. There are two forms of vitamin D supplementations available cynically for supplementation viz. Cholecalciferol (D3) and ergocalciferol (D2). These supplementations are available in various doses like 400, 600, 800, 1000, 2000, 5000, 10000, 50000, 60000 IU capsules, powder or tablets. In some countries they are available in Injectable form as well.
Vitamin d supplementation can be done by any of following regimen depending up on patient factors like severity, patients’ absorptive ability, compliance or clinical manifestations:
Initially 60000 IU of D2 or D3 once a week for 6-8 weeks (about 2 months) then 800 IU per day
1000 IU of D2 or D3 per day
600-800 IU of D2 or D3 per day
10000 to 60000 IU per day for malabsorption disorders depending upon severity of malabsorption and deficiency
In some cases, vitamin D metabolites like calcidiol or calcitriol or dihydrotachysterol may be used for treatment of vitamin D deficiency. Another modality of vitamin D deficiency treatment is artificial exposure to UVB (ultraviolet B) light.
Is vitamin D3 the same as vitamin D 25 hydroxyvitamin D3?
25-hydroxyvitamin D3 is one of the inactive forms of the vitamin D which is found in blood and its value is measured to check for vitamin D deficiency.
Is vitamin D same as D3 or D2?
Vitamin D has two forms, which are vitamin D2 and D3. The source of vitamin D3 is skin and animal foods where as vitamin D2 is found in plant sources.
Do we overdose on vitamin D?
Toxic dose of vitamin D supplementation is not clear though tolerable upper limit is set. For children above 9 years and adults, the largest upper limit is 4000 IU (100mcg) per day, while that for children it lower. Following symptoms might be seen if vitamin D toxicity or overdose occurs:
Decreased appetite
Weight loss
Irregular heartbeat
What should I do if I think I lack vitamin D?
If you think you have vitamin D deficiency you need to visit your doctor and he will ask you some questions about symptoms and signs of vitamin D deficiency. He may order some tests to confirm if you have vitamin D deficiency. After the reports he will treat it depending upon multiple factors and personalized treatment plan for you. He will also ask you for follow-up to confirm the correction of the deficiency, relief of symptoms, and help you with future prevention of the same condition. A repeat check of vitamin D level can usually be done after 3-4 months of supplementation intake. You can also book an appointment with us if you think you have vitamin D deficiency or any health problem. Use the contact us button or the chat box below. Thank you for reading.
Based on Nelson Textbook of Pediatrics, 21st Edition and recent updates
Table of Contents
febrile seizures definition
Introduction
Febrile seizures are the most common seizure disorder in childhood, occurring in association with fever but without evidence of central nervous system infection or acute electrolyte imbalance. They represent a benign, age-limited condition affecting genetically predisposed children.
Epidemiology
Age group: 6 months to 5 years (peak: 12–18 months)
Incidence: ~2–5% of children in most populations
Recurrence rate: ~30–35% after first episode; higher in early onset (<1 year)
Family history: Positive in up to 25–40% cases, suggesting genetic susceptibility
Definition (Nelson)
A febrile seizure is defined as a seizure accompanied by fever (>38°C or 100.4°F), without evidence of CNS infection, metabolic abnormality, or a history of afebrile seizures.
Classification
1. Simple Febrile Seizure (SFS)
Generalized tonic-clonic in onset
Duration <15 minutes
Occurs once in 24 hours
No postictal neurological deficit
2. Complex (Atypical) Febrile Seizure (CFS)
Focal onset or focal features during/post seizure
Duration >15 minutes
Recurrent within 24 hours
May have postictal weakness (Todd’s paresis)
3. Febrile Status Epilepticus (FSE)
Febrile seizure lasting >30 minutes (or series lasting ≥30 min without full recovery)
Requires urgent management
Etiopathogenesis
Genetic predisposition:
Polygenic inheritance; linkage to FEB1–FEB11 loci (e.g., FEB4 on 5q14–q15)
GABRG2, SCN1A gene mutations implicated (especially when overlapping with GEFS+)
Fever and cytokine response:
Elevated IL-1β, IL-6, and TNF-α lower seizure threshold
Rapid temperature rise rather than peak temperature triggers seizure
Immature brain excitability:
Age-dependent increased neuronal excitability due to GABA-A receptor subunit composition and immature synaptic inhibition
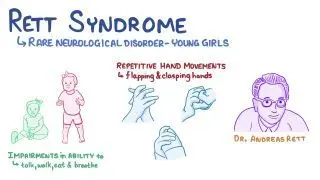
Rett syndrome is a neurodevelopmental disorder that primarily affects girls, characterized by normal early development followed by regression of acquired skills, especially speech and purposeful hand movements, with onset typically between 6–18 months of age.
Etiology
Genetic cause: Mutation in MECP2 gene (methyl-CpG-binding protein 2) on the X chromosome (Xq28)
Inheritance: Usually sporadic (de novo); rarely familial
Pathophysiology: Dysfunction of MECP2 protein → abnormal brain maturation and synaptic development
Epidemiology
Affects 1 in 10,000–15,000 female births
Lethal in males (most do not survive infancy unless mosaic or XXY)
Clinical Features
Phases of Disease
Early Onset (6–18 months)
Normal development initially
Gradual loss of interest in surroundings
Loss of purposeful hand skills
Deceleration of head growth (acquired microcephaly)
Rapid Destructive Phase (1–4 years)
Loss of speech and purposeful hand use
Stereotyped hand movements: hand-wringing, washing, clapping, or mouthing
Gait ataxia, truncal apraxia
Autistic-like behavior
Plateau Phase (2–10 years)
Some improvement in social interaction and eye contact
Hypothyroidism is a clinical state resulting from deficiency of thyroid hormone production or action, leading to a generalized slowing of metabolic processes.
It may be:
Congenital (Neonatal) – present at birth.
Acquired (Childhood) – develops later due to autoimmune, iatrogenic, or other causes.
2. Classification
A. Based on Level of Defect
Type
Site of Defect
TSH
T4/T3
Primary
Thyroid gland
↑
↓
Secondary
Pituitary
↓/N
↓
Tertiary
Hypothalamus
↓/N
↓
Peripheral (Resistance)
Target tissue
N/↑
N/↑
B. Based on Onset
Congenital hypothyroidism (CH)
Acquired hypothyroidism
3. Epidemiology
CH: ~1 in 2,000–4,000 live births.
More common in females.
Acquired form common in older children/adolescents, often autoimmune (Hashimoto’s).
thyroid gland
4. Etiology
A. Congenital Hypothyroidism
Thyroid dysgenesis (80–85%)
Agenesis, ectopy, or hypoplasia.
Usually sporadic.
Dyshormonogenesis (10–15%)
Inborn errors of thyroid hormone synthesis (autosomal recessive).
Leishmaniasis — MD Pediatrics Note (Based on Nelson Textbook of Pediatrics)
Table of Contents(toc)
Introduction
Leishmaniasis is a spectrum of protozoal diseases caused by Leishmania species, transmitted by the bite of infected female phlebotomine sandflies.
cutaneous leishmaniasis
Disease manifestations depend on the species involved and the host immune response.
Major clinical forms:
Visceral leishmaniasis (VL / kala-azar)
Cutaneous leishmaniasis (CL)
Mucocutaneous leishmaniasis (MCL)
Etiology and Classification
Form
Causative Species
Geographic Distribution
Visceral
L. donovani, L. infantum (chagasi)
South Asia, East Africa, Latin America
Cutaneous
L. tropica, L. major, L. mexicana, L. braziliensis
Middle East, Africa, Americas
Mucocutaneous
L. braziliensis complex
Central & South America
Epidemiology
Endemic in >80 countries; affects poor, rural populations.
Vectors:Phlebotomus (Old World), Lutzomyia (New World).
Reservoirs: Humans (L. donovani), dogs, rodents.
Transmission: Sandfly bite, rarely congenital or via transfusion.
Phlebotomus
Pathogenesis
Inoculation of promastigotes → engulfed by macrophages → transform into amastigotes → intracellular multiplication → spread to RES (liver, spleen, bone marrow).
Disease severity depends on cell-mediated immunity (CMI).
Episodic (Viral) Wheeze vs. Multiple Trigger Wheeze
A Clinically Oriented Review for the Practicing Pediatrician
Based on Nelson Textbook of Pediatrics (21st ed.) | Kendig’s Disorders of the Respiratory Tract in Children (9th ed.) | AAP & IAP-NAPCON Official Resources
1. Introduction
Wheezing in preschool children (0–5 years) is one of the most common reasons for pediatric consultation and hospital admission worldwide. It is now well established that ‘preschool wheeze’ is not a single disease but a heterogeneous group of phenotypes with distinct pathophysiology, natural history, and responses to therapy. The two most clinically useful and validated phenotypes—recognized in both the Nelson Textbook of Pediatrics and major international guidelines—are:
Episodic (Viral) Wheeze (EVW): wheezing episodes triggered exclusively by viral respiratory infections, with complete resolution between episodes.
Multiple Trigger Wheeze (MTW): wheezing triggered by multiple stimuli including viruses, aeroallergens, exercise, cold air, tobacco smoke, and emotional stimuli, with symptoms also occurring between discrete episodes.
This classification, initially proposed by Brand et al. and incorporated into the PRACTALL Consensus Report (2008) of the European Academy of Allergy and Clinical Immunology (EAACI) and the American Academy of Allergy, Asthma and Immunology (AAAAI), is now endorsed by the American Academy of Pediatrics (AAP) and the Indian Academy of Pediatrics (IAP) / National Asthma Consensus Group (NACG).
2. Epidemiology
According to Nelson Textbook of Pediatrics (21st edition, Chapter 169: Wheezing in Infants and Children), approximately 30–40% of all children will experience at least one wheezing episode in the first three years of life, yet fewer than one-third of these will develop persistent asthma. Data from the Tucson Children’s Respiratory Study (TCRS), cited prominently in Nelson, delineates three early wheezing trajectories:
Transient early wheezers: viral-triggered, remit by age 6; low atopic burden.
Non-atopic wheezers (EVW phenotype): episode-only wheeze; best aligned with EVW.
IgE-associated persistent wheezers (MTW/Asthma phenotype): atopic sensitization, family history, persistent into school age.
The IAP NAPCON 2019 Consensus Statement on Childhood Asthma notes that in South Asian children, including India and Nepal, the prevalence of preschool wheeze is significant, often complicated by high pollution exposure and early sensitization to house dust mite and cockroach allergens, features that shift the phenotype toward MTW.
3. Pathophysiology
3.1 Episodic (Viral) Wheeze
As described in Nelson (Chapter 169) and Kendig’s Disorders of the Respiratory Tract in Children (9th edition, Chapter 38), EVW is predominantly mediated by:
Rhinovirus (RV) and respiratory syncytial virus (RSV) — the principal triggers in children <3 years.
Neutrophilic airway inflammation: transient bronchial inflammation during the acute episode, with restoration of normal airway architecture between episodes. Unlike classical asthma, eosinophilic infiltration is typically absent or minimal.
Small airway mechanics: infants have a high ratio of airway resistance due to anatomically smaller caliber airways, making them more susceptible to luminal obstruction from viral-induced mucosal edema and secretions.
Immune dysregulation: reduced interferon-γ (IFN-γ) and impaired Th1 responses to RV have been demonstrated, contributing to prolonged viral shedding and exaggerated bronchospasm.
No persistent structural remodeling: between episodes, lung function is typically normal and there is no evidence of airway remodeling or eosinophilic inflammation.
3.2 Multiple Trigger Wheeze
MTW pathophysiology, as detailed in both Nelson and Kendig’s, resembles that of classic atopic asthma:
Eosinophilic airway inflammation: persistent even during asymptomatic intervals, with elevated fractional exhaled nitric oxide (FeNO).
Th2-skewed immune response: elevated IgE, IL-4, IL-5, IL-13; mast cell and eosinophil activation with allergen exposure.
Airway hyperresponsiveness (AHR): demonstrable on methacholine or exercise challenge, and persisting between symptomatic episodes.
Early sensitization: specific IgE to house dust mite (Dermatophagoides pteronyssinus), cockroach, Alternaria, or other regional allergens is frequently demonstrable by age 2–3 years.
Structural remodeling: subepithelial fibrosis and smooth muscle hypertrophy develop over time if left inadequately treated.
4. Clinical Features and Diagnosis
4.1 History
Nelson (21st ed., Chapter 169) and AAP Clinical Practice Guidelines for Asthma (2020 Update) recommend a detailed history focusing on:
Trigger identification: exclusive viral triggers (EVW) vs. multiple triggers including allergens, exercise, cold air, irritants (MTW).
Inter-episodic symptoms: nocturnal cough, exercise-induced wheeze, or persistent cough between viral episodes strongly suggests MTW.
Atopic comorbidities: personal history of eczema, allergic rhinitis; food allergy.
Family history: parental asthma/atopy increases the Asthma Predictive Index (API) score, supporting MTW/asthma phenotype.
Environmental history: tobacco smoke exposure, cooking fuel, pet ownership, damp housing — relevant especially per IAP guidelines for South Asian settings.
4.2 Asthma Predictive Index (API)
The modified API (mAPI), described in Nelson and endorsed by the AAP, is a validated tool to identify preschool wheezers likely to develop persistent asthma (MTW phenotype). A positive mAPI in a child with ≥3 wheezing episodes in the past year has a positive predictive value of ~80% for asthma at school age.
Digital clubbing, persistent hyperinflation, failure to thrive — suggest alternative diagnoses (cystic fibrosis, primary ciliary dyskinesia, structural airway anomalies).
Normal examination between episodes — expected in EVW; persistent wheeze or hyperinflation between episodes raises suspicion for MTW or alternative pathology.
4.4 Investigations
Kendig’s (9th ed., Chapter 38) and AAP Guidelines recommend the following investigations based on clinical context:
Spirometry (≥5–6 years): reversible airflow obstruction (post-bronchodilator FEV1 improvement ≥12%) supports MTW/asthma; may be normal in EVW.
Skin prick testing / Specific IgE: aeroallergen sensitization supports MTW phenotype; recommended in children with positive mAPI or recurrent MTW.
Complete blood count: peripheral eosinophilia (≥4%) is a minor API criterion.
Chest radiograph: to exclude structural anomalies, foreign body, or consolidation; not routinely needed for wheeze per AAP guidelines.
FeNO measurement: elevated (>25 ppb) supports eosinophilic airway inflammation (MTW/asthma); not universally available but referenced in Nelson and Kendig’s.
Bronchoscopy / BAL: reserved for diagnostically challenging cases; mentioned in Kendig’s for evaluation of structural/anatomic causes of wheeze.
5. Comparative Overview: EVW vs. MTW
Table 1 summarizes the key distinguishing features of the two preschool wheeze phenotypes.
Table 1. Episodic Viral Wheeze vs. Multiple Trigger Wheeze — Comparative Features
Per AAP Clinical Practice Guidelines (2020) and Nelson (Chapter 169), acute management is phenotype-independent and follows standard bronchodilator therapy:
Short-Acting Beta-2 Agonists (SABA): salbutamol (albuterol) 2.5–5 mg via nebulizer, or 2–4 puffs via spacer and face mask every 20 minutes for 3 doses in severe episodes. First-line therapy for all preschool wheeze.
Ipratropium bromide: may be added for moderate-to-severe exacerbations; reduces hospitalization when combined with salbutamol.
Systemic corticosteroids: oral prednisolone (1–2 mg/kg/day, max 40 mg, for 3–5 days) for moderate-to-severe exacerbations. Per the AAP, short courses do not significantly affect adrenal function or growth in children.
Supplemental oxygen: titrate to maintain SpO2 ≥94% (AAP target); SpO2 ≥95% per IAP-NAPCON 2019.
Hospitalization criteria: SpO2 <92% on room air, severe respiratory distress (HR >60/min in infants), inability to maintain oral feeds, poor response to initial bronchodilators.
7.2 Preventive/Controller Therapy
This is where the phenotype distinction critically guides management:
7.2.1 Episodic (Viral) Wheeze
Per Nelson, Kendig’s, and AAP Guidelines:
Continuous ICS: NOT routinely recommended for EVW. Multiple RCTs (including the PEAK and MIST trials cited in Nelson) show no significant reduction in episode frequency or severity with continuous low-dose ICS in non-atopic preschool wheezers.
Intermittent/episodic ICS: high-dose ICS at the onset of a viral URTI (e.g., budesonide 400 mcg/day or fluticasone 200 mcg/day for 7–10 days) may reduce episode severity in selected children, though evidence remains inconsistent across trials.
Montelukast: episodic use at onset of wheeze shows modest benefit in some studies (Bisgaard et al., NEJM, cited in Nelson); may be considered for children with 3 or more episodes per year.
Bronchodilator reliever therapy: salbutamol as needed during episodes. Continuous reliever use between episodes is not indicated in pure EVW.
Avoidance: passive smoking cessation, hand hygiene, daycare modifications to reduce viral exposure.
7.2.2 Multiple Trigger Wheeze
Per Nelson, Kendig’s, AAP (2020), and IAP-NAPCON (2019):
Low-dose ICS: first-line preventer therapy. Budesonide 100–200 mcg/day or fluticasone propionate 100 mcg/day (BDP-equivalent). Initiate when diagnosis of MTW/persistent asthma is established.
Montelukast: may be used as an alternative to ICS in mild MTW or as add-on therapy in moderate MTW. IAP-NAPCON recognizes its role given high house dust mite sensitization in the South Asian context.
Medium-dose ICS: step up to 200–400 mcg/day (budesonide equivalent) if low-dose ICS fails to achieve symptom control after 6–8 weeks.
LABA addition: for children ≥5 years with inadequate control on medium-dose ICS, salmeterol or formoterol can be added. Not approved or recommended for children <4 years as monotherapy.
Allergen avoidance: mattress/pillow encasements, HEPA filtration, pet removal — strongly recommended by AAP and IAP for sensitized children with MTW.
Allergen Immunotherapy (AIT): subcutaneous or sublingual AIT for house dust mite-sensitized children with MTW/asthma is recommended in international guidelines and endorsed in IAP-NAPCON for appropriate candidates ≥5 years.
Omalizumab: anti-IgE therapy; approved for moderate-to-severe persistent allergic asthma in children ≥6 years; referenced in Nelson and AAP guidelines for refractory MTW/asthma with high IgE and allergen sensitization.
7.3 Step-Therapy Summary
Table 2. Stepwise Treatment Approach for EVW and MTW
Step
EVW Management
MTW Management
Acute
SABA (salbutamol) via spacer/nebulizer; oral prednisolone for moderate-severe
SABA; oral/systemic corticosteroids; consider early ICS step-up
Preventer
Not routinely indicated; trial ICS only if frequent/severe episodes (≥3/year)
Low-dose ICS (e.g., budesonide 100–200 mcg/day) as first-line preventer
Step-up
Episodic ICS at onset of URTI (intermittent therapy); montelukast episodic use
Increase ICS dose; add montelukast or LABA (≥5 yr); consider specialist referral
Monitoring
Symptom diary; reassess trigger pattern at each visit
Nebulizers are not superior to pMDI+spacer for acute bronchodilation and carry infection transmission risk in healthcare settings. Both AAP and IAP recommend prioritizing spacer-based delivery.
8. Monitoring and Follow-Up
Nelson, AAP (2020 Expert Panel Report 3 Update), and IAP-NAPCON recommend the following monitoring framework:
Review diagnosis every 3–6 months: re-evaluate whether phenotype has shifted from EVW to MTW as the child grows.
Assess symptom control using validated tools: \Childhood Asthma Control Test (C-ACT) for children ≥4 years; parent-report tools for younger children.
Spirometry when developmentally feasible (≥5 years): monitor FEV1, FVC, and FEV1/FVC ratio at each visit.
Reassess trigger profile at each visit: new aeroallergen sensitization, school exposures, change in environment.
Monitor growth: height and weight percentile; ICS at low doses does not significantly affect final adult height per Nelson; monitor with medium-to-high doses.
Adherence and inhaler technique: check at every visit; poor technique is the most common cause of apparent treatment failure per AAP.
Consider step-down: if well-controlled for ≥3 months, cautiously step down therapy, reassessing trigger pattern.
9. Prognosis and Natural History
The TCRS and birth cohort studies cited in Nelson provide the most robust data on prognosis:
EVW (Transient wheeze): ~60% of preschool wheezers remit by 6 years of age. These children, corresponding to the EVW phenotype, generally have normal lung function at school age. The absence of atopic sensitization, normal lung function between episodes, and non-positive API predict favorable outcome.
MTW (Persistent/Asthma phenotype): ~40% of preschool wheezers continue to wheeze at school age. Risk factors for persistence include: positive mAPI, maternal asthma, early sensitization to aeroallergens, frequent episodes in the first 3 years, male sex, and exposure to high-dose indoor allergens.
Lung function trajectory: Lung function deficits, if present at age 6 years in the MTW group, tend to track into adult life and are associated with increased risk of COPD in adulthood (“early origins of adult lung disease” concept, cited in Nelson and Kendig’s).
South Asian context (IAP): earlier sensitization to perennial allergens (HDM, cockroach), higher pollution burden, and lower vitamin D levels may confer worse outcomes in the MTW phenotype in Indian children, as noted in IAP-NAPCON 2019.
10. Special Clinical Situations
10.1 The “Overlap” Child
Many children present with features of both EVW and MTW, especially between ages 2–4 years. Nelson recommends using the mAPI as a practical decision aid in such cases. If the mAPI is positive, treat as MTW (initiate regular ICS); if negative, manage as EVW (episodic/as-needed therapy).
10.2 Very Young Infants (<12 months)
Wheezing in infants under 12 months is most commonly due to bronchiolitis (RSV) and should not be classified as EVW or MTW. Per AAP Clinical Practice Guideline for the Diagnosis, Management, and Prevention of Bronchiolitis (2014, reaffirmed 2020), bronchodilators are not recommended for infants with bronchiolitis. ICS and systemic steroids are similarly not recommended in this age group for acute bronchiolitis.
10.3 COVID-19 and Respiratory Viruses
The AAP has issued guidance noting that SARS-CoV-2 infection in young children may trigger wheezing episodes similar to other viral URTI triggers in EVW. Standard asthma action plans should include COVID-19 as a potential EVW trigger; ICS should not be stopped during COVID-19 illness in MTW/asthma patients.
10.4 Vaccination
Both AAP and IAP recommend annual influenza vaccination for all children with recurrent wheezing (EVW or MTW), as influenza is a significant trigger for severe exacerbations. Pneumococcal vaccination per national immunization schedules is also recommended.
11. Parent and Caregiver Education
AAP and IAP emphasize that education is a cornerstone of management:
Provide written Asthma Action Plan (AAP template available at healthychildren.org) for all children with recurrent wheeze.
Educate on symptom recognition: early signs of exacerbation (nocturnal cough, reduced exercise tolerance, increased rescue inhaler use).
Inhaler technique training at every visit; video demonstrations and teach-back methods are recommended by AAP.
Environmental control counseling: tobacco smoke, allergen avoidance, mold reduction, pet dander management.
Address caregiver anxiety: explain phenotype, natural history, and that EVW does not inevitably become asthma.
Emphasize adherence to preventive therapy in MTW: parents often reduce ICS doses prematurely when symptoms improve.
12. Key Clinical Takeaways
Phenotype matters: Distinguish EVW from MTW at every clinical encounter; this distinction drives preventive therapy decisions.
mAPI guides therapy: A positive mAPI in a high-frequency preschool wheezer indicates MTW/asthma phenotype and justifies early ICS therapy.
ICS is not universal: Continuous ICS is not recommended for pure EVW; reserve for MTW or EVW with frequent/severe episodes.
Trigger profile shapes management: Allergen sensitization testing is indicated when MTW is suspected; AIT may be indicated in sensitized children ≥5 years.
Phenotypes are dynamic: Reassess at every visit; EVW may evolve to MTW as atopic sensitization develops.
Guideline resources: Use AAP (healthychildren.org, aappublications.org) and IAP-NAPCON (iapindia.org) official resources for updated local guidance.
Exclude mimics: Always consider structural, infectious, and congenital causes of recurrent wheeze, especially in children <12 months or with atypical features.
References
Primary Textbook References:
Kliegman RM, St. Geme JW, Blum NJ, et al. Nelson Textbook of Pediatrics, 21st Edition. Philadelphia: Elsevier; 2020. Chapter 169: Wheezing in Infants and Young Children; Chapter 170: Asthma.
Wilmott RW, Deterding R, Li A, et al. Kendig’s Disorders of the Respiratory Tract in Children, 9th Edition. Philadelphia: Elsevier; 2019. Chapter 38: Wheezing in Infancy and Early Childhood; Chapter 39: Asthma in the Pediatric Patient.
AAP Official Resources:
American Academy of Pediatrics. Clinical Practice Guideline for the Diagnosis, Evaluation, and Management of Childhood Asthma. Pediatrics. 2020;145(3):e20193432. Available at: https://publications.aap.org
American Academy of Pediatrics. Clinical Practice Guideline: The Diagnosis, Management, and Prevention of Bronchiolitis. Pediatrics. 2014;134(5):e1474-e1502. Reaffirmed 2020. Available at: https://publications.aap.org
American Academy of Pediatrics. Asthma Action Plan templates and parent education resources. HealthyChildren.org. Available at: https://www.healthychildren.org
IAP Official Resources:
Indian Academy of Pediatrics, National Asthma Consensus Group (NAPCON). IAP-NAPCON Consensus Statement on Childhood Asthma 2019. Indian Pediatrics. 2020;57(1):42–58. Available at: https://www.indianpediatrics.net
Indian Academy of Pediatrics. IAP Standard Treatment Guidelines: Bronchial Asthma in Children. 2022. Available at: https://www.iapindia.org
Landmark Studies and Consensus Documents (cited in Nelson/Kendig’s):
Brand PL, Baraldi E, Bisgaard H, et al. Definition, assessment and treatment of wheezing disorders in preschool children: an evidence-based approach. European Respiratory Journal. 2008;32(4):1096–1110. [PRACTALL Consensus Report, cited in Nelson 21e and Kendig’s 9e]
Martinez FD, Wright AL, Taussig LM, et al. Asthma and wheezing in the first six years of life: The Group Health Medical Associates. New England Journal of Medicine. 1995;332(3):133–138. [Tucson Children’s Respiratory Study, cited in Nelson 21e]
National Asthma Education and Prevention Program (NAEPP). Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma. National Heart, Lung, and Blood Institute (NHLBI). 2007 (Updated 2020). Available at: https://www.nhlbi.nih.gov
Global Initiative for Asthma (GINA). Difficult-to-Treat and Severe Asthma in Adolescent and Adult Patients: A GINA Pocket Guide. 2023. [Referenced in Nelson and Kendig’s for management framework]
Malnutrition: Complications, Assessment, and Prevention
Malnutrition is a serious public health concern affecting individuals of all ages, particularly children in low-resource settings. It can lead to both acute and chronic complications, impacting survival, growth, and overall health.
Acute Complications of Malnutrition
A helpful mnemonic for remembering the main acute complications is “Shieldeded”:
Sugar deficiency / Hypoglycemia – Low blood sugar levels can lead to lethargy, seizures, and even coma.
Hypothermia – Impaired thermoregulation increases vulnerability to cold stress.
Infection – Reduced immunity predisposes to frequent and severe infections.
Electrolyte disorder – Commonly includes imbalances in sodium, potassium, and magnesium.
Dehydration – Often due to diarrhea or inadequate fluid intake.
Deficiency of vitamins and minerals – Leads to a range of specific deficiency syndromes (e.g., anemia, rickets, night blindness).
Next we will discuss 10 essential steps in the management of malnutrition (Severe Acute Malnutrition – SAM) based on standard WHO guidelines.
Management of Malnutrition (SAM) – 10 Steps
Step
Management
Key Actions
Timeline
1
Treat/Prevent Hypoglycemia
Give glucose immediately, start frequent feeds
Immediately (within first hours)
2
Treat/Prevent Hypothermia
Keep child warm, kangaroo care
Immediately & ongoing (first 24 hrs)
3
Treat/Prevent Dehydration
Use ReSoMal, careful rehydration
First 24 hours
4
Correct Electrolyte Imbalance
Give potassium, magnesium, restrict sodium
First 1–2 days
5
Treat Infections
Start broad-spectrum antibiotics
Immediately (Day 1)
6
Correct Micronutrient Deficiencies
Vitamin A, zinc, folate (avoid iron initially)
Day 1 onward
7
Start Cautious Feeding
Begin F-75 diet (stabilization phase)
First 2–7 days
8
Achieve Catch-up Growth
Switch to F-100 or RUTF
After stabilization (Day 7+)
9
Provide Sensory Stimulation
Play therapy, emotional care
Throughout treatment
10
Prepare for Follow-up
Nutrition education, immunization, monitoring
Before discharge & after recovery
Chronic Complications of Malnutrition
Untreated or prolonged malnutrition can result in chronic health problems:
Pseudotumour cerebri – Raised intracranial pressure without a brain tumor, causing headaches and visual disturbances.
Anthropometry – Measurement of weight, height, mid-upper arm circumference (MUAC), and growth charts.
Biochemical markers – Blood tests to assess nutrient levels and detect deficiencies.
Clinical evaluation – Physical examination for signs of malnutrition.
Dietary evaluation – Analysis of food intake patterns and adequacy.
Epidemiological assessment – Community-based data to identify at-risk populations.
Prevention of Malnutrition: GOBIFFF Strategy
The GOBIFFF approach is widely promoted for prevention:
G – Growth monitoring
O – Oral rehydration solution (ORS) use
B – Breastfeeding promotion
I – Immunization coverage
F – Family planning
F – Female education
F – Feeding improvement (appropriate complementary feeding)
Follow-up in Malnutrition
Monitoring recovery is crucial for preventing relapse:
Initial follow-up: At 2 weeks, 1 month, and 3 months after starting treatment.
Long-term follow-up: Every 3 months thereafter until the Z-score is greater than –1.
Assessment of Physical Growth Schedule
Monthly for children under 1 year
Every 2 months for ages 1–2 years
Every 3 months for ages 3–5 years
Conclusion
Malnutrition remains preventable through early detection, community education, and targeted interventions. A combination of clinical vigilance and public health measures can ensure healthier growth and development in children worldwide.
Gastrointestinal (GI) Bleeding in Children: High-Yield Overview
Table of Contents(toc)
Gastrointestinal (GI) Bleeding in Children: High-Yield Overview
GI bleeding in children is classified into upper and lower sources. Understanding the common causes and their relative prevalence helps in timely diagnosis and management.
Upper GI Bleeding(More Common)
Esophagitis, Gastritis, Duodenitis – 30–40%
Most frequent causes; often associated with infections, NSAIDs, or stress.
Gastroesophageal Reflux Disease (GERD) – 20–30%
Chronic reflux can lead to mucosal damage and bleeding.
Peptic Ulcer Disease – 10–20%
Associated with H. pylori, stress, or NSAIDs.
Esophageal Varices – 5–10%
Seen in children with chronic liver disease or portal hypertension.
Mallory-Weiss Tear – ~5%
Mucosal tear due to forceful vomiting.
Coagulopathies / Bleeding Disorders – 2–5%
Underlying bleeding diathesis may present with GI hemorrhage.
Foreign Body Ingestion (with mucosal injury) – <5%
Particularly in toddlers; bleeding due to mucosal erosion or ulceration.
Lower GI Bleeding
Anal Fissures – 30–40%
Most common cause in infants and toddlers; associated with hard stools.
Infectious Colitis / Gastroenteritis – 20–25%
Caused by bacterial or viral pathogens, often with diarrhea.
Juvenile Polyps – 10–15%
Benign but can cause painless rectal bleeding in young children.
Meckel’s Diverticulum – 5–10%
Congenital anomaly; may bleed due to ectopic gastric mucosa.
Inflammatory Bowel Disease (IBD) – 5–10%
Includes Crohn’s and ulcerative colitis; chronic inflammation leads to bleeding.
Intussusception – 2–5%
Often presents with “currant jelly” stools and abdominal pain.
Henoch-Schönlein Purpura (HSP) – 1–5%
Small vessel vasculitis; GI involvement can cause bleeding and pain.
Here is a quick-reference table summarizing the common causes of GI bleeding in children, categorized by location and including approximate prevalence:
Common Causes of GI Bleeding in Children
Upper GI Bleeding
Prevalence
Esophagitis / Gastritis / Duodenitis
30–40%
Gastroesophageal Reflux Disease (GERD)
20–30%
Peptic Ulcer Disease
10–20%
Esophageal Varices
5–10%
Mallory-Weiss Tear
~5%
Coagulopathies / Bleeding Disorders
2–5%
Foreign Body Ingestion (with mucosal injury)
<5%
Lower GI Bleeding
Prevalence
Anal Fissures
30–40%
Infectious Colitis / Gastroenteritis
20–25%
Juvenile Polyps
10–15%
Meckel’s Diverticulum
5–10%
Inflammatory Bowel Disease (IBD)
5–10%
Intussusception
2–5%
Henoch-Schönlein Purpura (HSP)
1–5%
Stay Connected with Dr. Chaitanya Joshi, MD
YouTube Channel
Watch health videos, tips, and updates from Dr. Chaitanya MD.































")








